Human Gene PSMD12 (ENST00000356126.8) from GENCODE V44
Description: Homo sapiens proteasome 26S subunit, non-ATPase 12 (PSMD12), transcript variant 1, mRNA. (from RefSeq NM_002816) RefSeq Summary (NM_002816): The 26S proteasome is a multicatalytic proteinase complex with a highly ordered structure composed of 2 complexes, a 20S core and a 19S regulator. The 20S core is composed of 4 rings of 28 non-identical subunits; 2 rings are composed of 7 alpha subunits and 2 rings are composed of 7 beta subunits. The 19S regulator is composed of a base, which contains 6 ATPase subunits and 2 non-ATPase subunits, and a lid, which contains up to 10 non-ATPase subunits. Proteasomes are distributed throughout eukaryotic cells at a high concentration and cleave peptides in an ATP/ubiquitin-dependent process in a non-lysosomal pathway. An essential function of a modified proteasome, the immunoproteasome, is the processing of class I MHC peptides. This gene encodes a non-ATPase subunit of the 19S regulator. A pseudogene has been identified on chromosome 3. Alternatively spliced transcript variants encoding different isoforms have been found for this gene. [provided by RefSeq, Oct 2015]. Gencode Transcript: ENST00000356126.8 Gencode Gene: ENSG00000197170.10 Transcript (Including UTRs) Position: hg38 chr17:67,337,916-67,366,577 Size: 28,662 Total Exon Count: 11 Strand: - Coding Region Position: hg38 chr17:67,340,843-67,366,519 Size: 25,677 Coding Exon Count: 11
ID:PSD12_HUMAN DESCRIPTION: RecName: Full=26S proteasome non-ATPase regulatory subunit 12; AltName: Full=26S proteasome regulatory subunit RPN5; AltName: Full=26S proteasome regulatory subunit p55; FUNCTION: Acts as a regulatory subunit of the 26S proteasome which is involved in the ATP-dependent degradation of ubiquitinated proteins. SUBUNIT: Component of the PA700 complex. SIMILARITY: Belongs to the proteasome subunit p55 family. SIMILARITY: Contains 1 PCI domain. SEQUENCE CAUTION: Sequence=AAH65826.1; Type=Erroneous initiation;
The RNAfold program from the Vienna RNA Package is used to perform the secondary structure predictions and folding calculations. The estimated folding energy is in kcal/mol. The more negative the energy, the more secondary structure the RNA is likely to have.
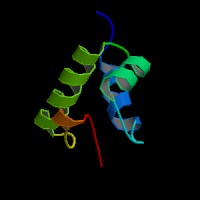
ModBase Predicted Comparative 3D Structure on O00232
Front
Top
Side
The pictures above may be empty if there is no ModBase structure for the protein. The ModBase structure frequently covers just a fragment of the protein. You may be asked to log onto ModBase the first time you click on the pictures. It is simplest after logging in to just click on the picture again to get to the specific info on that model.
Orthologous Genes in Other Species
Orthologies between human, mouse, and rat are computed by taking the best BLASTP hit, and filtering out non-syntenic hits. For more distant species reciprocal-best BLASTP hits are used. Note that the absence of an ortholog in the table below may reflect incomplete annotations in the other species rather than a true absence of the orthologous gene.
Gene Ontology (GO) Annotations with Structured Vocabulary
Biological Process: GO:0016579 protein deubiquitination GO:0043161 proteasome-mediated ubiquitin-dependent protein catabolic process GO:0043312 neutrophil degranulation GO:0043687 post-translational protein modification