Human Gene RGR (ENST00000359452.9) from GENCODE V44
Description: Homo sapiens retinal G protein coupled receptor (RGR), transcript variant 1, mRNA. (from RefSeq NM_002921) RefSeq Summary (NM_001012720): This gene encodes a putative retinal G-protein coupled receptor. The gene is a member of the opsin subfamily of the 7 transmembrane, G-protein coupled receptor 1 family. Like other opsins which bind retinaldehyde, it contains a conserved lysine residue in the seventh transmembrane domain. The protein acts as a photoisomerase to catalyze the conversion of all-trans-retinal to 11-cis-retinal. The reverse isomerization occurs with rhodopsin in retinal photoreceptor cells. The protein is exclusively expressed in tissue adjacent to retinal photoreceptor cells, the retinal pigment epithelium and Mueller cells. This gene may be associated with autosomal recessive and autosomal dominant retinitis pigmentosa (arRP and adRP, respectively). Alternative splicing results in multiple transcript variants encoding different isoforms. [provided by RefSeq, Jul 2008]. Gencode Transcript: ENST00000359452.9 Gencode Gene: ENSG00000148604.15 Transcript (Including UTRs) Position: hg38 chr10:84,245,079-84,259,960 Size: 14,882 Total Exon Count: 7 Strand: + Coding Region Position: hg38 chr10:84,245,091-84,258,639 Size: 13,549 Coding Exon Count: 7
ID:RGR_HUMAN DESCRIPTION: RecName: Full=RPE-retinal G protein-coupled receptor; FUNCTION: Receptor for all-trans- and 11-cis-retinal. Binds preferentially to the former and may catalyze the isomerization of the chromophore by a retinochrome-like mechanism. INTERACTION: Q96EK5:KIAA1279; NbExp=2; IntAct=EBI-745818, EBI-744150; SUBCELLULAR LOCATION: Membrane; Multi-pass membrane protein. TISSUE SPECIFICITY: Preferentially expressed at high levels in the retinal pigment epithelium (RPE) and Mueller cells of the neural retina. PTM: Covalently binds all-trans- and 11-cis-retinal. DISEASE: Defects in RGR are the cause of retinitis pigmentosa type 44 (RP44) [MIM:613769]. RP44 is a retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well. SIMILARITY: Belongs to the G-protein coupled receptor 1 family. Opsin subfamily. WEB RESOURCE: Name=Mutations of the RGR gene; Note=Retina International's Scientific Newsletter; URL="http://www.retina-international.org/files/sci-news/rgrmut.htm"; WEB RESOURCE: Name=GeneReviews; URL="http://www.ncbi.nlm.nih.gov/sites/GeneTests/lab/gene/RGR";
The RNAfold program from the Vienna RNA Package is used to perform the secondary structure predictions and folding calculations. The estimated folding energy is in kcal/mol. The more negative the energy, the more secondary structure the RNA is likely to have.
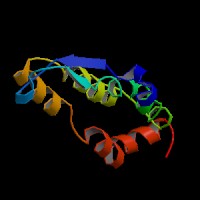
ModBase Predicted Comparative 3D Structure on P47804
Front
Top
Side
The pictures above may be empty if there is no ModBase structure for the protein. The ModBase structure frequently covers just a fragment of the protein. You may be asked to log onto ModBase the first time you click on the pictures. It is simplest after logging in to just click on the picture again to get to the specific info on that model.
Orthologous Genes in Other Species
Orthologies between human, mouse, and rat are computed by taking the best BLASTP hit, and filtering out non-syntenic hits. For more distant species reciprocal-best BLASTP hits are used. Note that the absence of an ortholog in the table below may reflect incomplete annotations in the other species rather than a true absence of the orthologous gene.