VCF+tabix Track Format
Variant Call Format (VCF) is a flexible and extendable line-oriented text format developed by the 1000 Genomes Project (now maintained by the GA4GH) for releases of single nucleotide variants, indels, copy number variants and structural variants discovered by the project. When a VCF file is compressed and indexed using tabix, and made web-accessible, the Genome Browser is able to fetch only the portions of the file necessary to display items in the viewed region. This makes it possible to display variants from files that are so large that the connection to UCSC would time out when attempting to upload the whole file to UCSC. Both the VCF file and its tabix index file remain on your web-accessible server (http, https, or ftp), not on the UCSC server. UCSC temporarily caches the accessed portions of the files to speed up interactive display. If you do not have access to a web-accessible server and need hosting space for your VCF files, please see the Hosting section of the Track Hub Help documentation.
The UCSC tools support VCF versions 3.3 and greater.
If you have VCF trio data, you may be interested in formatting your track as a Phased Trios track as described below.
VCF Format
VCF is an all-purpose format for defining variants of all types: SNVs, CNVs and translocations relative to a reference assembly. It can annotate all the variants in an individual as well as a population. Typically, a VCF file is too large to load directly into a custom track on the Browser and must be loaded as binary tabix-indexed file as described below. The full specification of VCF is found in documents on github.
Here is a look at an example from that file showing a few rows of data for three samples. Details and descriptions of the data fields are in the .pdf. The data here can be pasted directly into hg18 human assembly on the Genome Browser. We have added two lines at the top of the entry that are not in the official example to make the display work in the Browser.
Note that the first data field, identifying the chromosome, is in official VCF format which does not include the "chr" usually associated with Genome Browser chrom names. Either version will work in the Browser.
track type=vcf name="vcf example" description="three samples in a vcf" db=hg18 visibility="full" browser position chr20:1-1306000 ##fileformat=VCFv4.2 ##fileDate=20090805 ##source=myImputationProgramV3.1 ##reference=file:///seq/references/1000GenomesPilot-NCBI36.fasta ##contig=<ID=20,length=62435964,assembly=B36,md5=f126cdf8a6e0c7f379d618ff66beb2da,species="Homo sapiens",taxonomy=x> ##phasing=partial ##INFO=<ID=NS,Number=1,Type=Integer,Description="Number of Samples With Data"> ##INFO=<ID=DP,Number=1,Type=Integer,Description="Total Depth"> ##INFO=<ID=AF,Number=A,Type=Float,Description="Allele Frequency"> ##INFO=<ID=AA,Number=1,Type=String,Description="Ancestral Allele"> ##INFO=<ID=DB,Number=0,Type=Flag,Description="dbSNP membership, build 129"> ##INFO=<ID=H2,Number=0,Type=Flag,Description="HapMap2 membership"> ##FILTER=<ID=q10,Description="Quality below 10"> ##FILTER=<ID=s50,Description="Less than 50% of samples have data"> ##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype"> ##FORMAT=<ID=GQ,Number=1,Type=Integer,Description="Genotype Quality"> ##FORMAT=<ID=DP,Number=1,Type=Integer,Description="Read Depth"> ##FORMAT=<ID=HQ,Number=2,Type=Integer,Description="Haplotype Quality"> #CHROM POS ID REF ALT QUAL FILTER INFO FORMAT NA00001 NA00002 NA00003 20 14370 rs6054257 G A 29 PASS NS=3;DP=14;AF=0.5;DB;H2 GT:GQ:DP:HQ 0|0:48:1:51,51 1|0:48:8:51,51 1/1:43:5:.,. 20 17330 . T A 3 q10 NS=3;DP=11;AF=0.017 GT:GQ:DP:HQ 0|0:49:3:58,50 0|1:3:5:65,3 0/0:41:3 20 1110696 rs6040355 A G,T 67 PASS NS=2;DP=10;AF=0.333,0.667;AA=T;DB GT:GQ:DP:HQ 1|2:21:6:23,27 2|1:2:0:18,2 2/2:35:4 20 1230237 . T . 47 PASS NS=3;DP=13;AA=T GT:GQ:DP:HQ 0|0:54:7:56,60 0|0:48:4:51,51 0/0:61:2 20 1234567 microsat1 GTC G,GTCT 50 PASS NS=3;DP=9;AA=G GT:GQ:DP 0/1:35:4 0/2:17:2 1/1:40:3
Generating a VCF track
The typical workflow for generating a VCF custom track is this:
- If you haven't done so already, download and build the tabix and bgzip programs. Test your installation by running tabix with no command-line arguments; it should print a brief usage message. For help with tabix, please contact the samtools-help mailing list (tabix is part of the samtools project).
- Create VCF or convert another format to VCF. Items must be sorted by genomic position.
-
Compress your .vcf file using the
bgzipprogram:
For more information about thebgzip my.vcfbgzipcommand, run it with no arguments to display the usage message. -
Create a tabix index file (
.tbior.csi) for the bgzip-compressed VCF (.vcf.gz). By default, the tabix command appends .tbi to the my.vcf.gz filename, creating a binary index file named my.vcf.gz.tbi with which genomic coordinates can quickly be translated into file offsets in my.vcf.gz:
The tabix (tabix -p vcf my.vcf.gz.tbi) and BAI index formats can handle individual chromosomes up to 512 Mbp (2^29 bases) in length. If your input file contains data lines with start or end positions greater than 512 Mbp, you will need to use a CSI (.csi) index instead.tabix --csi -p vcf my.vcf.gz -
Move both the compressed VCF file (my.vcf.gz) and index file
(my.vcf.gz.tbi or my.vcf.gz.csi) to an http, https, or ftp location. Note that
the Genome Browser looks for an index file with the same URL as the VCF file with the .tbi or .csi
suffix added.
If your hosting site does not use the filename as the URL link, you will have to specifically call the location of this .vcf.tbi/csi index file with thebigDataIndexkeyword. You can read more about bigDataIndex in the TrackDb Database Definition page. -
Construct a custom track using a single
track line. The basic version of the track line will look
something like this:
Again, in addition to http://myorg.edu/mylab/my.vcf.gz, the associated index file http://myorg.edu/mylab/my.vcf.gz.tbi must also be available at the same location. If the file is in a different location or uses a different filename, then use the bigDataIndex attribute in the track line to point to the index file.track type=vcfTabix name="My VCF" bigDataUrl=http://myorg.edu/mylab/my.vcf.gztrack type=vcfTabix name="My VCF" bigDataUrl=http://myorg.edu/mylab/my.vcf.gz bigDataIndex=http://myorg.edu/someOtherDirectory/myvcf.gz.tbi - Paste the custom track line into the text box in the custom track management page, click "submit" and view in the Genome Browser.
Parameters for VCF custom track definition lines
All options are placed in a single line separated by spaces (lines are broken only for readability here):
track type=vcfTabix bigDataUrl=http://...
hapClusterEnabled=true|false
hapClusterColorBy=altOnly|refAlt|base
hapClusterTreeAngle=triangle|rectangle
hapClusterHeight=N
applyMinQual=true|false minQual=Q
minFreq=F
name=track_label
description=center_label
visibility=display_mode
priority=priority
db=db maxWindowToDraw=N
chromosomes=chr1,chr2,... Note if you copy/paste the above example, you must remove the line breaks. Click here for a text version that you can paste without editing.
The track type and bigDataUrl are REQUIRED:
type=vcfTabix bigDataUrl=http://myorg.edu/mylab/my.vcf.gz The remaining settings are OPTIONAL. Some are specific to VCF:
hapClusterEnabled true|false # if file has phased genotypes, sort by local similarity
hapClusterColorBy altOnly|refAlt|base # coloring scheme, default altOnly, conditional on hapClusterEnabled
hapClusterTreeAngle triangle|rectangle # draw leaves as < or [, default <, conditional on hapClusterEnabled
hapClusterHeight N # height of track in pixels, default 128, conditional on hapClusterEnabled
applyMinQual true|false # if true, don't display items with QUAL < minQual; default false
minQual Q # minimum value of Q column to display item, conditional on applyMinQual
minFreq F # minimum minor allele frequency to display item; default 0.0 Other optional settings are not specific to VCF, but relevant:
name track label # default is "User Track"
description center label # default is "User Supplied Track"
visibility squish|pack|full|dense|hide # default is hide (will also take numeric values 4|3|2|1|0)
priority N # default is 100
db genome database # e.g. hg19 for Human Feb. 2009 (GRCh37)
maxWindowToDraw N # don't display track when viewing more than N bases
chromosomes chr1,chr2,... # track contains data only on listed reference assembly sequences Phased Trio format
The vcfPhasedTrio track type is available for users whose VCF contains genotype data from one to three individuals. The underlying VCF follows the standard VCF format as described above, with the added caveat that there must be GENOTYPE columns for each of the individuals present. An example of the trio display is shown below for the 1000 Genomes Trio track on Human/GRCh38:
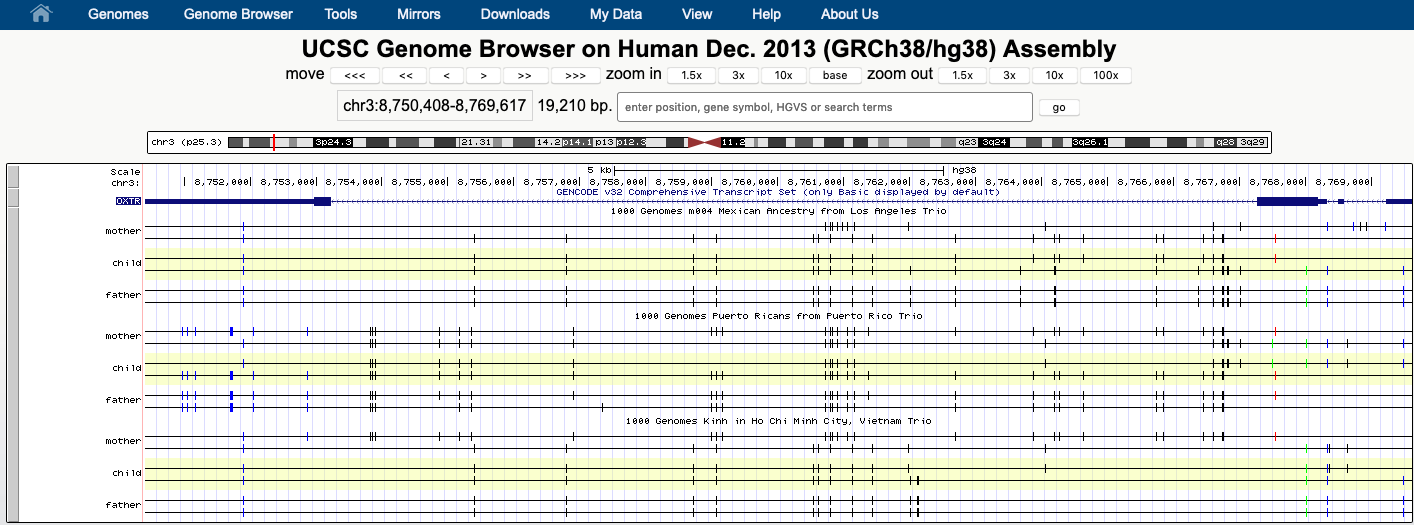
Unlike a regular genome browser track, Trio tracks display the genome variants of each individual as two haplotypes; SNPs, small insertions and deletions are mapped to each haplotype based on the phasing information of the VCF file. Each haplotype is displayed on two separate, horizontal black lines across the browser window. Each variant is drawn as a vertical dash. Homozygous variants will show two identical dashes on both haplotype lines. Phased heterozygous variants are placed on one of the haplotype lanes and unphased heterozygous variants are displayed in the area between the two haplotype lines.
Follow the steps for a normal VCF file, including moving the file to a web accessible location and generating a tabix index file, then use the following required vcfPhasedTrio trackDb settings to view the trio display:
type vcfPhasedTrio # The track type is required and must be "vcfPhasedTrio"
bigDataUrl http://url.to.vcfFile # The bigDataUrl is required
vcfChildSample GT ID|alias # the Genotype column ID of the "child" sample, with an optional "|" followed by a human readable alias for the ID
There are also two optional settings for vcfPhasedTrio tracks:
vcfParentSamples GT ID1|alias1,GT ID2|alias2 # comma separated (no spaces) list of the "parent" samples, with optional aliases
vcfUseAltSampleNames GT ID # Use the aliases in the display by default instead of the Genotype column ID
Other optional settings are not specific to VCF, but relevant:
maxWindowToDraw N # don't display track when viewing more than N bases
chromosomes chr1,chr2,... # track contains data only on listed reference assembly sequences Examples
Example #1
In this example, you will create a custom track for an indexed VCF file that is already on a public server — variant calls generated by the 1000 Genomes Project. The line breaks inserted here for readability must be removed before submitting the track line:
browser position chr21:33,034,804-33,037,719
track type=vcfTabix name="VCF Example One" description="VCF Ex. 1: 1000 Genomes phase 1 interim SNVs"
chromosomes=chr21 maxWindowToDraw=200000
db=hg19 visibility=pack
bigDataUrl=http://genome.ucsc.edu/goldenPath/help/examples/vcfExample.vcf.gz The "browser" line above is used to view a small region of chromosome 21 with variants from the .vcf.gz file.
Note if you copy/paste the above example, you must remove the line breaks (or, click here for a text version that you can paste without editing).
Paste the "browser" line and "track" line into the custom track management page for the human assembly hg19 (Feb. 2009), then click the "submit" button. On the following page, click the "chr21" link in the custom track listing to view the VCF track in the Genome Browser.
Example #2
In this example, you will create compressed, indexed VCF from an existing VCF text file. First, save this VCF file -- vcfExampleTwo.vcf -- to your machine. Perform steps 1 and 3-7 in the workflow described above, but substitute vcfExampleTwo.vcf for my.vcf. On the custom track management page, click the "add custom tracks" button if necessary and make sure that the genome is set to "Human" and the assembly is set to "Feb. 2009 (hg19) " before pasting the track line and submitting. Remember to remove the line breaks that have been added to the track line for readability (or, click here for a text version that you can paste without editing):
track type=vcfTabix name="VCF Example Two" bigDataUrl=http://myorg.edu/mylab/vcfExampleTwo.vcf.gz
description="VCF Ex. 2: More variants from 1000 Genomes" visibility=pack db=hg19 chromosomes=chr21
browser position chr21:33,034,804-33,037,719
browser pack snp132CommonExample #3
In this example, you will load a hub that has VCF data described in a hub's trackDb.txt file. First, navigate to the Basic Hub Quick Start Guide and review an introduction to hubs.
Visualizing VCF files in hubs involves creating three text files: hub.txt, genomes.txt, and trackDb.txt. The browser is passed a URL to the top-level hub.txt file that points to the related genomes.txt and trackDb.txt files. The trackDb.txt file contains stanzas for each track that outlines the details and type of each track to display, such as these lines for a VCF file located at the bigDataUrl location:
track vcf1
bigDataUrl http://hgdownload.gi.ucsc.edu/gbdb/hg19/1000Genomes/ALL.chr21.integrated_phase1_v3.20101123.snps_indels_svs.genotypes.vcf.gz
#Note: there is a corresponding fileName.vcf.gz.tbi in the same above directory
shortLabel chr21 VCF example
longLabel This chr21 VCF file is an example from the 1000 Genomes Phase 1 Integrated Variant Calls Track
type vcfTabix
visibility dense
Note: there is now a useOneFile on hub setting that allows the hub
properties to be specified in a single file. More information about this setting can be found on the
Genome Browser User Guide.
Here is a direct link to the trackDb.txt file to see more information about this example hub, and below is a direct link to visualize the hub in the browser, where this example VCF file displays in dense mode alongside the other tracks in this hub. You can find more Track Hub VCF display options on the Track Database (trackDb) Definition Document page.
http://genome.ucsc.edu/cgi-bin/hgTracks?db=hg19&hubUrl=http://genome.ucsc.edu/goldenPath/help/examples/hubDirectory/hub.txt Sharing Your Data with Others
If you would like to share your VCF data track with a colleague, learn how to create a URL by looking at Example 6 on the custom tracks page.